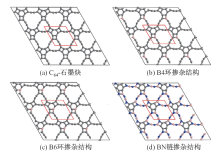
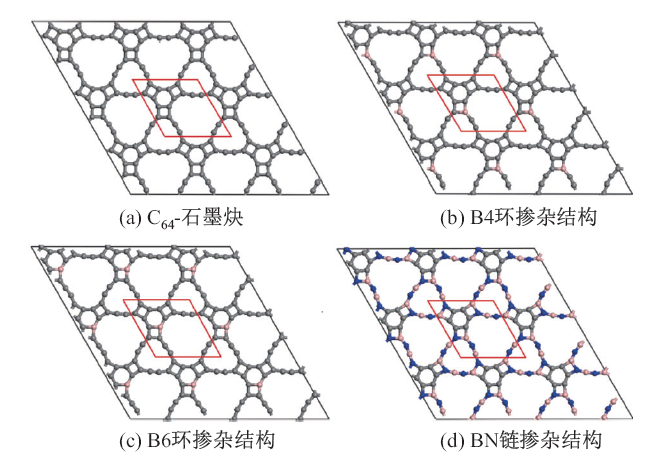
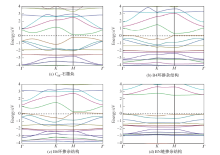
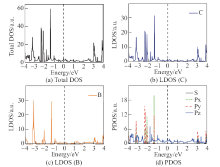
| [1] |
Novoselov K S, Geim A K, Morozov S V, et al. Electric field effect in atomically thin carbon films[J]. Science, 2004,306(5696):666-669.
pmid: 15499015
|
| [2] |
Neto A H C, Guinea F, Peres N M R, et al. The electronic properties of graphene[J]. Reviews of Modern Physics, 2009,81(1):109-162.
|
| [3] |
Nair R R, Blake P, Grigorenko A N, et al. Fine structure constant defines visual transparency of graphene[J]. Science, 2008,320(5881):1308.
doi: 10.1126/science.1156965
pmid: 18388259
|
| [4] |
Kuloglu A F, Sarikavak-Lisesivdin B, Lisesivdin S B, et al. First-principles calculations of Pd-terminated symmetrical armchair graphene nanoribbons[J]. Computational Materials Science, 2013,68:18-22.
|
| [5] |
Wu M, Cao C, Jiang J Z. Light non-metallic atom (B, N, O and F)-doped graphene: a first-principles study[J]. Nanotechnology, 2010,21(50):505202.
pmid: 21098927
|
| [6] |
Wei D, Liu Y, Wang Y, et al. Synjournal of N-doped graphene by chemical vapor deposition and its electrical properties[J]. Nano Letters, 2009,9(5):1752-1758.
pmid: 19326921
|
| [7] |
Sheng Z H, Gao H L, Bao W J, et al. Synjournal of boron doped graphene for oxygen reduction reaction in fuel cells[J]. Journal of Materials Chemistry, 2011,22(2):390-395.
|
| [8] |
Panchakarla L S, Subrahmanyam K S, Saha S K, et al. Synjournal, structure, and properties of boron and nitrogen doped graphene[J]. Advanced Materials, 2009,21(46):4726-4730.
|
| [9] |
Baughman R H, Eckhardt H, Kertesz M. Structure-property predictions for new planar forms of carbon: layered phases containing sp 2 and sp atoms[J]. The Journal of Chemical Physics, 1987,87(11):6687-6699.
doi: 10.1063/1.453405
|
| [10] |
Li G, Li Y, Liu H, et al. Architecture of graphdiyne nanoscale films[J]. Chemical Communications, 2010,46(19):3256-3258.
doi: 10.1039/b922733d
pmid: 20442882
|
| [11] |
Zhang Y Y, Pei Q X, Wang C M. Mechanical properties of graphynes under tension: a molecular dynamics study[J]. Applied Physics Letters, 2012,101(8):081909.
doi: 10.1063/1.4747719
|
| [12] |
Long M, Tang L, Wang D, et al. Electronic structure and carrier mobility in graphdiyne sheet and nanoribbons: theoretical predictions[J]. Acs Nano, 2011,5(4):2593-2600.
doi: 10.1021/nn102472s
pmid: 21443198
|
| [13] |
Ma S Y, Zhang M, Sun L Z, et al. High-temperature behavior of monolayer graphyne and graphdiyne[J]. Carbon, 2016,99(11):547-555.
|
| [14] |
Zhang J J, Xin Z H, Zhang J H, et al. Molecular dynamics study on the stability and properties of alpha-CGeyne[J]. Acta Physica Sinica, 2014,63(20):207303.
|
| [15] |
Liu J, Xin Z, Yan X, et al. Structural, phononic and electronic properties of Ge-doped $\gamma$-graphynes: a first-principles study[J]. Solid State Communications, 2017,258:38-44.
|
| [16] |
Yan X, Xin Z H, Zhang J J, et al. Molecular dynamics study on the structure and properties of silicon-graphdiyne[J]. Acta Physica Sinica, 2013,62(23):238101.
|
| [17] |
Singh N B, Bhattacharya B, Sarkar U. A first principle study of pristine and BN-doped graphyne family[J]. Structural Chemistry, 2014,25(6):1695-1710.
|
| [18] |
Kang B, Shi H, Wang F F, et al. Importance of doping site of B, N, and O in tuning electronic structure of graphynes[J]. Carbon, 2016,105:156-162.
|
| [19] |
Yun J, Zhang Z, Yan J, et al. First-principles study of B or Al-doping effect on the structural, electronic structure and magnetic properties of $\gamma$-graphyne[J]. Computational Materials Science, 2015,108(12):147-152.
|
| [20] |
Kim H, Kim Y, Kim J, et al. Computational searching for new stable graphyne structures and their electronic properties[J]. Carbon, 2016,98:404-410.
|
| [21] |
Song Q, Wang B, Deng K, et al. Graphenylene, a unique two-dimensional carbon network with nondelocalized cyclohexatriene units[J]. Journal of Materials Chemistry C, 2013,1(1):38-41.
|
| [22] |
Lu H G, Li S D. Two-dimensional carbon allotropes from graphene to graphyne[J]. Journal of Materials Chemistry C, 2013,1(23):3677-3680.
|
 )
)