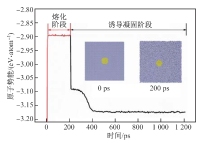
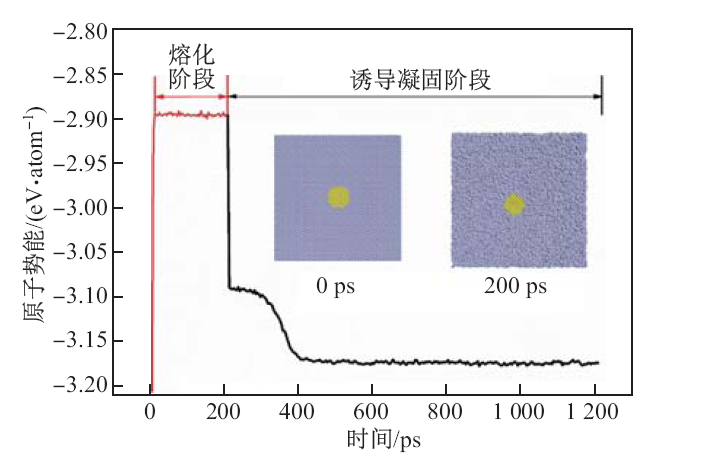
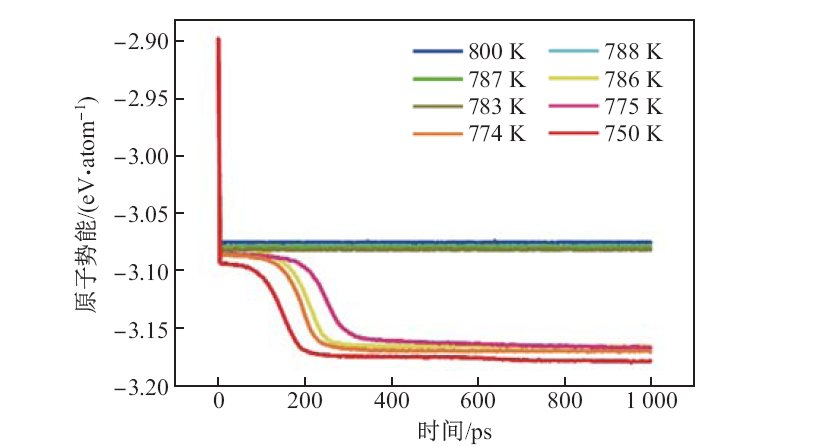
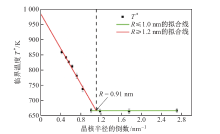
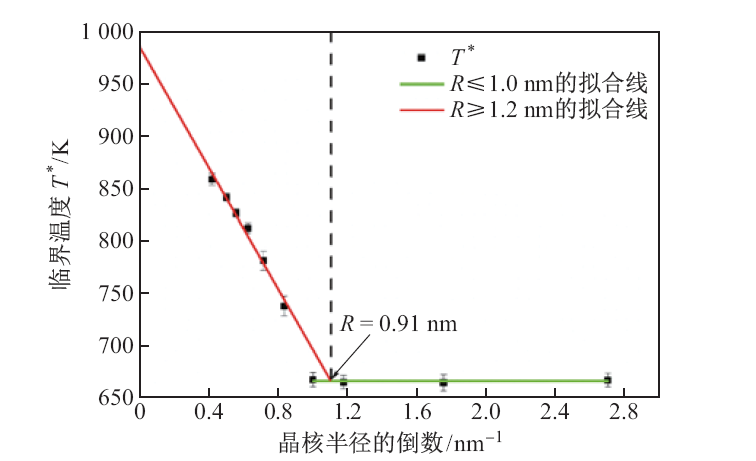
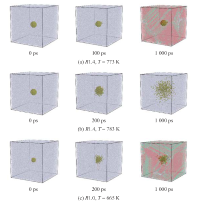
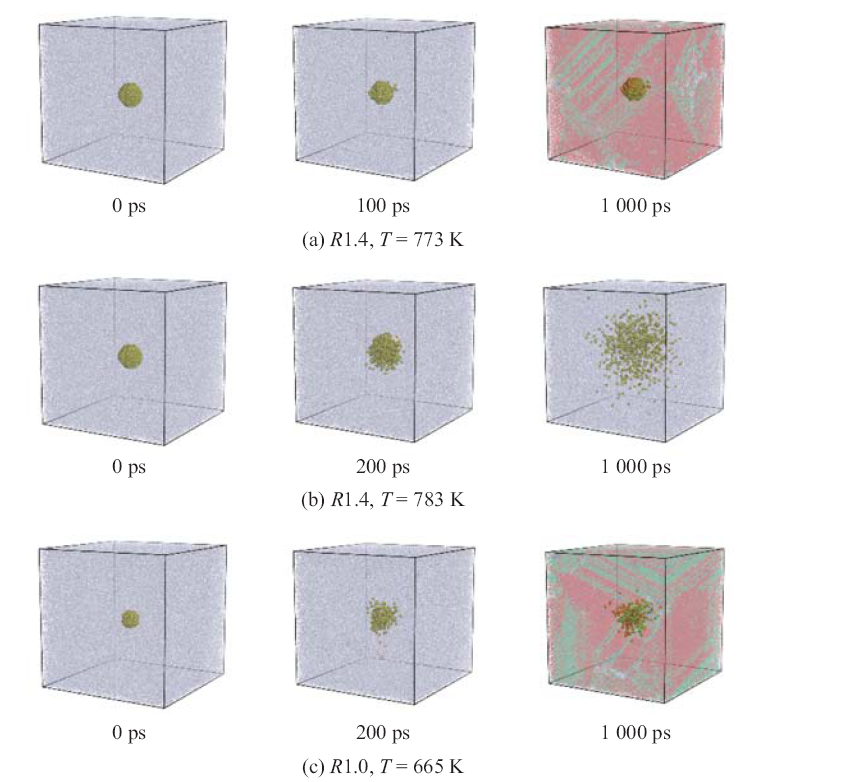
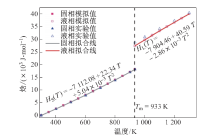
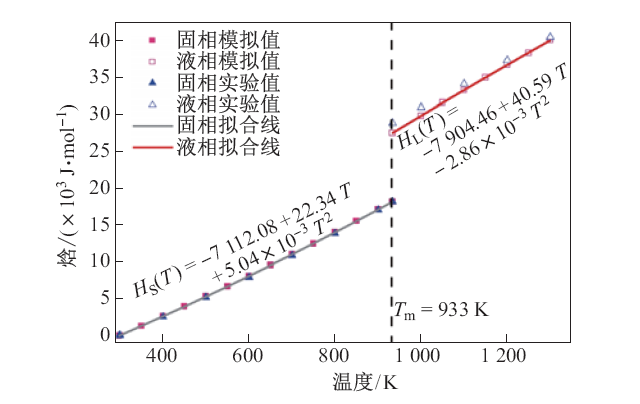
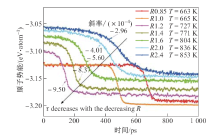
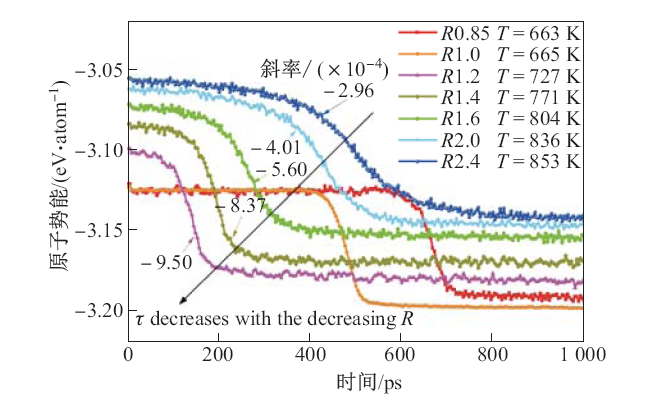
| [1] |
Turnbull D . Formation of crystal nuclei in liquid metals[J]. Journal of Applied Physics, 1950,21(10):1022-1028.
doi: 10.1063/1.1699435
|
| [2] |
Espinosa J R, Vega C, Valeriani C , et al. Seeding approach tocrystal nucleation[J]. Journal of Chemical Physics, 2016,144(3):1-10.
|
| [3] |
Bai X M, Li M . Calculation of solid-liquid interfacial freeenergy: a classical nucleation theory based approach[J]. Journal ofChemical Physics, 2006,124(12):1-12.
doi: 10.1063/1.2184315
pmid: 16599718
|
| [4] |
Broughton J Q, Gilmer G H . Molecular dynamics investigation ofthe crystal-fluid interface. VI. Excess surface free energies ofcrystal-liquid systems[J]. Journal of Chemical Physics, 1986,84(10):5759-5768.
doi: 10.1063/1.449884
|
| [5] |
Hoyt J J, Asta M, Karma A . Method for computing the anisotropyof the solid-liquid interfacial free energy[J]. Physical ReviewLetters, 2001,86(24):5530-5533.
doi: 10.1103/PhysRevLett.86.5530
pmid: 11415293
|
| [6] |
Shibuta Y, Suzuki T . A molecular dynamics study of the phasetransition in BCC metal nanoparticles[J]. Journal of ChemicalPhysics, 2008,129(14):1-10.
doi: 10.1063/1.2991435
pmid: 19045129
|
| [7] |
Watanbe Y, Shibuta Y, Suzuki T . A molecular dynamics study ofthermodynamic and kinetic properties of solid-liquid interface forBCC iron[J]. ISIJ International, 2010,50(8):1158-1164.
doi: 10.2355/isijinternational.50.1158
|
| [8] |
Hashimoto R, Shibuta Y, Suzuki T . Estimation of solid-liquidinterfacial energy from Gibbs-Thomson effect: a molecular dynamicsstudy[J]. ISIJ International, 2011,51(10):1664-1667.
|
| [9] |
Xia Y, Li C H, Luan Y W , et al. Molecular dynamics studies onthe correlation of undercoolability and thermophysical properties ofliquid Ni-Al alloys[J]. Computational Materials Science, 2016,1112(1):383-394.
|
| [10] |
Wu Y Q, Shen T, Lu X M , et al. Solidification of liquid Fe withembedded homogeneous solid Fe nanoparticles from molecular dynamicssimulations[J]. Acta Physico-Chimica Sinica, 2013,29(2):245-249.
|
| [11] |
Plimpton S . Fast parallel algorithms for short-range moleculardynamics[J]. Journal of Computational Physics, 1995,117(1):1-19.
|
| [12] |
Mendelev M I, Han S, Srojovitz D J , et al. Development of newinteratomic potentials appropriate for crystalline and liquid iron[J]. Philosophical Magazine, 2003,83(35):3977-3994.
|
| [13] |
Jiang Y W, Luo J, Wu Y Q . The validation and preference amongdifferent EAM potentials to describe the solid-liquid transition ofaluminum[J]. Modelling and Simulation in Materials Science andEngineering, 2017,25(4):1-13.
|
| [14] |
Li R, Wu Y Q, Xiao J J . The nucleation process and the roles ofstructure and density fluctuations in supercooled liquid Fe[J]. Journal of Chemical Physics, 2014,140(3):1-11.
doi: 10.1063/1.4861587
pmid: 25669396
|
| [15] |
Zhou H G, Lin X, Wang M , et al. Calculation of crystal-meltinterfacial free energies of FCC metals[J]. Journal of CrystalGrowth, 2013,366(3):82-87.
|
| [16] |
Liu J, Dong H B . Molecular dynamics calculation ofthermodynamic properties of iron solidification[J]. IOP ConferenceSeries: Materials Science and Engineering, 2012,33(1):1-10.
|
| [17] |
Chase M W, Curnutt J L, Downey J R , et al. JANAF thermochemicaltables, 1982 supplement[J]. Journal of Physical and ChemicalReference Data, 1982,11(3):695-940.
|
| [18] |
Kelton K F . Crystal nucleation in liquids and glasses[J]. Solid State Physics, 1991,45(1):75-177.
|
| [19] |
Morris J R . Complete mapping of the anisotropic free energy ofthe crystal-melt interface in Al[J]. Physical Review B, 2002,66(14):1-7.
|
| [20] |
Morris J R, Mendelev M I, Srojovitz D J . A comparison ofcrystal-melt interfacial free energies using different Al potentials[J]. Journal of Non-Crystalline Solids, 2007,353(32):3565-3569.
|
 )
)